4 minutes de lecture
Suspecter une amylose cardiaque : première étape vers le diagnostic
Publié le dimanche 10 octobre 2021

Auteur :
Dr Diane Bodez
Centre Cardiologique du Nord
Saint-Denis
Les amyloses cardiaques sont aujourd’hui une part significative des cardiopathies hypertrophiques, et la première cause de cardiopathie restrictive (dans le monde occidental). Leur diagnostic fait aujourd’hui l’objet de recommandations codifiées, qui reposent en premier lieu sur la reconnaissance d’entités syndromiques où les amyloses cardiaques sont fréquemment rencontrées, associées à la présence de signes d’alerte, aussi appelés red flags.(1)
Circonstances diagnostiques évocatrices
Il existe quatre grands cadres cliniques où le cardiologue doit être particulièrement vigilant à la possibilité d’une amylose cardiaque : l’insuffisance cardiaque à FEVG préservée, l’hypertrophie myocardique, les troubles du rythme et de la conduction, et la sténose aortique, a fortiori si le patient est âgé de plus de 65 ans. Même si elles sont peu spécifiques, ce sont en effet les présentations cardiologiques les plus fréquentes, couvrant la quasi-totalité des premiers symptômes cardiaques des patients porteurs d’amylose.
Dans chacune de ces circonstances, certains critères additionnels devront renforcer la suspicion diagnostique :
- Contexte général : la présence d’une gammapathie monoclonale (type MGUS plus souvent que myélome) devra faire évoquer la possibilité d’une amylose AL ; l’existence d’antécédents familiaux d’amylose bien sûr, mais aussi d’insuffisance cardiaque ou de neuropathie périphérique est évocatrice d’amylose ATTRv
- Signes cardiaques :
- Bien que peu spécifiques, les aspects ECG le plus fréquemment rencontrés sont un voltage normal ou microvoltage discordant d’avec une hypertrophie échographique, un aspect de pseudo-onde Q en antéro-septal, des troubles de la conduction (BAV1 ou bloc de branche) ou du rythme (plus souvent supra-ventriculaire)
- L’échographie cardiaque retrouve typiquement un phénotype hypertrophique (épaisseur pariétale ≥ 12mm) ou restrictif avec aspect granité du myocarde (à apprécier en fonction des réglages techniques de l’échographe), un épaississement diffus touchant les parois ventriculaires, mais aussi les valves et le septum interatrial, et un épanchement péricardique généralement de faible abondance. L’analyse de la fonction systolique mettra en évidence une discordance entre une FEVG préservée jusqu’aux stades tardifs, et une altération plus précoce de la fonction longitudinale, avec altération des vitesses tissulaires et surtout du strain global longitudinal avec aspect typique en cocarde. L’association avec une sténose aortique est fréquente, particulièrement dans le cadre particulier des sténoses à bas gradient. Plusieurs systèmes de score diagnostique selon des critères échographiques ont été récemment proposés.(2)
- Les biomarqueurs cardiaques, exceptionnellement normaux, montrent une discordance entre un niveau disproportionnellement élevé de NT-proBNP pour le degré d’anomalies échographiques, et une élévation souvent en plateau de la troponine, témoignant de l’atteinte myocardique qui peut initialement orienter à tort vers une cardiopathie ischémique.
Les red flags à reconnaître
Dans les circonstances cliniques décrites précédemment, la présence d’un ou plusieurs des signes suivants doit particulièrement interpeller le cardiologue.
Plusieurs signes extra-cardiaques peuvent être rencontrés dans les amyloses systémiques, qui ne se limitent pas seulement aux amyloses AL : protéinurie même modérée, insuffisance rénale, macroglossie et ecchymoses péri-orbitaires (théoriquement pathognomonique d’amylose AL lorsqu’elles sont associées), neuropathie périphérique et/ou dysautonomie. L’existence d’un syndrome du canal carpien, a fortiori s’il est bilatéral chez un homme, est très évocatrice d’amylose ATTRwt, dont il précède souvent de plusieurs années les manifestations cardiaques, de même que le canal lombaire étroit, la rupture des tendons bicipitaux, ou la surdité.
Lors du bilan, certains éléments cardiologiques renforceront la suspicion diagnostique : présence d’une hypotension ou diminution des besoins en traitement antihypertenseur, aspects ECG et échocardiographiques évocateurs (cf. supra).
Une fois l’amylose suspectée, Il est alors relativement urgent de réaliser un bilan spécifique pour confirmer ou infirmer le diagnostic d’amylose, certaines formes représentant une urgence thérapeutique (amyloses AL).
Mémo : les différents types d’amyloses cardiaques
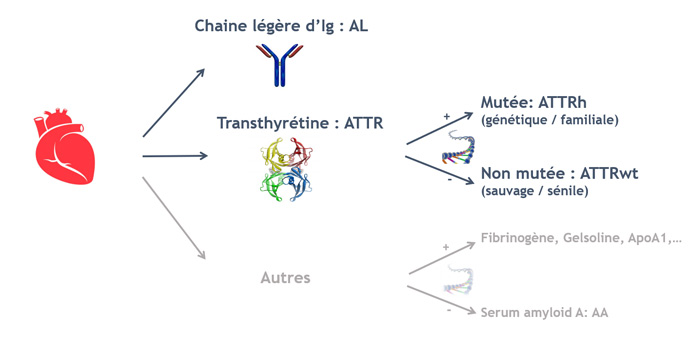
Figure 1 : les différents types d'amyloses cardiaques
AL : amylose à chaines légères ; ATTR : amylose à transthyrétine ; ATTRv : amylose à transthyrétine mutée ; ATTRwt : amylose à transthyrétine wild-type (sauvage)
Les amyloses cardiaques sont un groupe hétérogène de maladies, dont le point commun est l’accumulation progressive extra-cellulaire de fibrilles amyloïdes, dont le précurseur protéique définit le type d’amylose. Plusieurs dizaines de précurseurs protéiques existent, mais les plus fréquemment associés à une atteinte cardiaque sont :
- Les chaines légères d’immunoglobulines, produites en excès dans le cadre de gammapathies monoclonales, et responsables des amyloses AL,
- La transthyrétine, transporteur protéique produit par le foie, responsable des amyloses ATTR, qu’elles soient héréditaires (ATTRv) ou non (ATTR wild-type, ou ATTRwt, dites aussi sauvages ou séniles).
- Plus rarement la protéine SAA, présente dans les états inflammatoires chroniques, et responsable des amyloses AA.
- D’autres précurseurs protéiques, responsables d’amyloses génétiques rares, peuvent parfois être associés à une atteinte cardiaque (Apo A1, fibrinogène, etc.).
Références
- Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur J Heart Fail. avr 2021;23(4):512‑26.
- Boldrini M, Cappelli F, Chacko L, Restrepo-Cordoba MA, Lopez-Sainz A, Giannoni A, et al. Multiparametric Echocardiography Scores for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. avr 2020;13(4):909‑20.
Retrouvez l'intégralité du dossier spécial "Prise en charge de l'amylose cardiaque en 2021"
Ce contenu vous est proposé avec le soutien institutionnel de Pfizer Maladies Rares
Dans la même thématique



Articles les plus lus